Poznaj Narzędzie diamond Aligner
Zaawansowane oprogramowanie do błyskawicznego porównywania sekwencji białkowych
Czym jest diamond?
diamond to zaawansowane narzędzie open-source, stworzone do szybkiego dopasowywania sekwencji DNA i białek w obrębie ogromnych baz danych. Jego wydajność jest znacznie wyższa w porównaniu do tradycyjnych metod, takich jak BLASTX, dzięki zastosowaniu uproszczonego alfabetu aminokwasów oraz nowoczesnych technologii optymalizacyjnych, które przyspieszają analizę przy minimalnej utracie precyzji.
Narzędzie to stanowi szczególnie cenne wsparcie w badaniach metagenomicznych, analizie porównawczej sekwencji oraz w identyfikacji potencjalnych peptydów przeciwdrobnoustrojowych. Umożliwia efektywne przetwarzanie dużych zbiorów danych, jednak jego prawidłowe zastosowanie wymaga od użytkownika wiedzy w zakresie konfiguracji parametrów analizy, interpretacji wyników oraz dostępu do sprzętu o odpowiedniej wydajności obliczeniowej.
Jak Działa Porównywanie Sekwencji?
diamond porównuje każdą sekwencję znajdującą się w bazie referencyjnej z każdą sekwencją zawartą w bazie zapytania, obliczając procent identyczności pomiędzy parami sekwencji. Proces ten skutkuje wygenerowaniem bardzo dużej liczby indywidualnych porównań, których liczba rośnie wykładniczo wraz z wielkością analizowanych zbiorów danych. Dzięki temu możliwa jest szczegółowa i kompleksowa ocena stopnia podobieństwa sekwencji pomiędzy bazami danych.
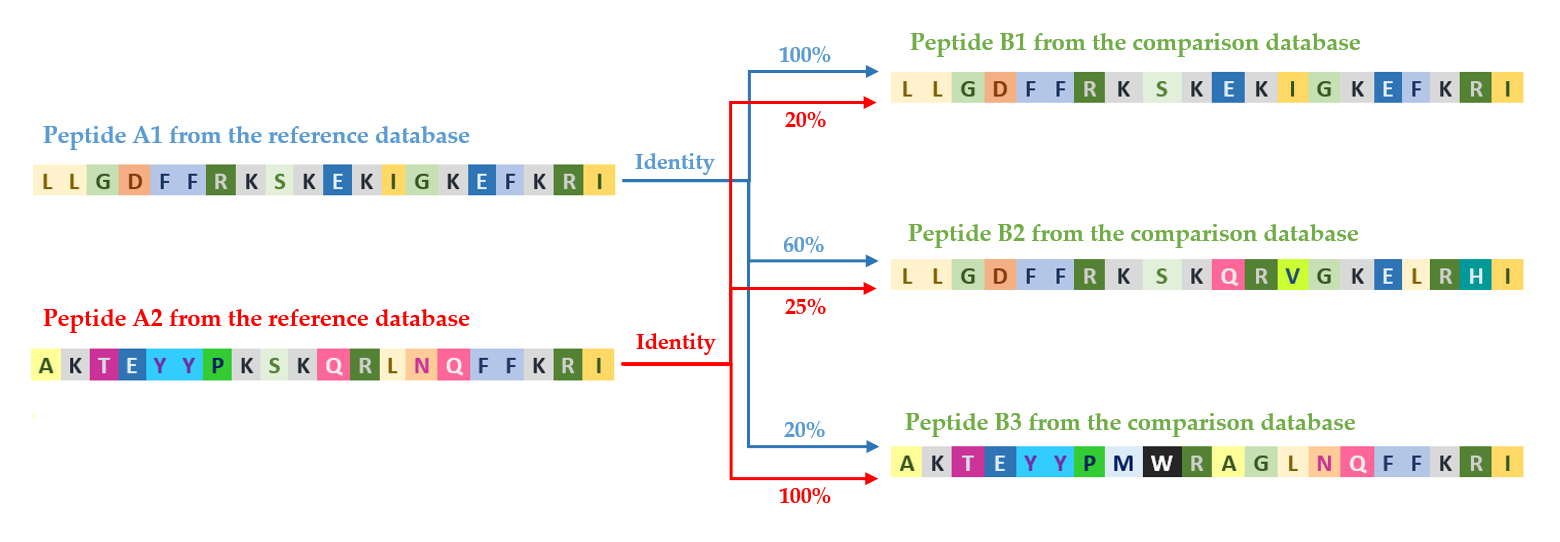
Rysunek przedstawia, jak sekwencje A1 i A2 z bazy referencyjnej są porównywane z sekwencjami B1, B2 i B3 z bazy porównawczej, co skutkuje określeniem procentowej identyczności dla każdej pary.
Kluczowe Cechy
Co sprawia, że diamond jest użytecznym narzędziem w bioinformatyce?
Niezwykła Szybkość
Do 20 000 razy szybszy niż BLASTX, co pozwala na analizę ogromnych zbiorów danych na standardowych serwerach.
Wysoka Czułość
Użycie zredukowanego alfabetu aminokwasów i technologii "spaced seed" zapewnia wysoką czułość przy minimalnej utracie precyzji.
Szerokie Zastosowanie
Idealny do analizy proteomów, projektów metagenomicznych i identyfikacji kandydatów na peptydy przeciwdrobnoustrojowe.
Bibliografia
Buchfink, B., Xie, C., & Huson, D.H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12, 59–60. https://doi.org/10.1038/nmeth.3176
Buchfink, B., Reuter, K., & Drost, H.G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods, 18, 366–368. https://doi.org/10.1038/s41592-021-01101-x
Marczak, B., Bocian, A., & Łyskowski, A. (2025). Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods. Molecules, 30, 1318. https://doi.org/10.3390/molecules30061318