Discover the diamond Aligner
Advanced software for lightning-fast protein sequence comparison
What is diamond?
diamond is an advanced open-source tool designed for rapid alignment of DNA and protein sequences against massive databases. Its performance is significantly higher compared to traditional methods like BLASTX, thanks to the use of a simplified amino acid alphabet and modern optimization technologies that accelerate analysis with minimal loss of precision.
This tool is particularly valuable for metagenomic research, comparative sequence analysis, and the identification of potential antimicrobial peptides. It allows for the efficient processing of large datasets, but its proper application requires the user to have knowledge of analysis parameter configuration, results interpretation, and access to hardware with adequate computational performance.
How does sequence comparison work?
diamond compares every sequence in the reference database with every sequence in the query database, calculating the percentage of identity between sequence pairs. This process generates a very large number of individual comparisons, the quantity of which grows exponentially with the size of the analyzed datasets. This enables a detailed and comprehensive assessment of the degree of sequence similarity between databases.
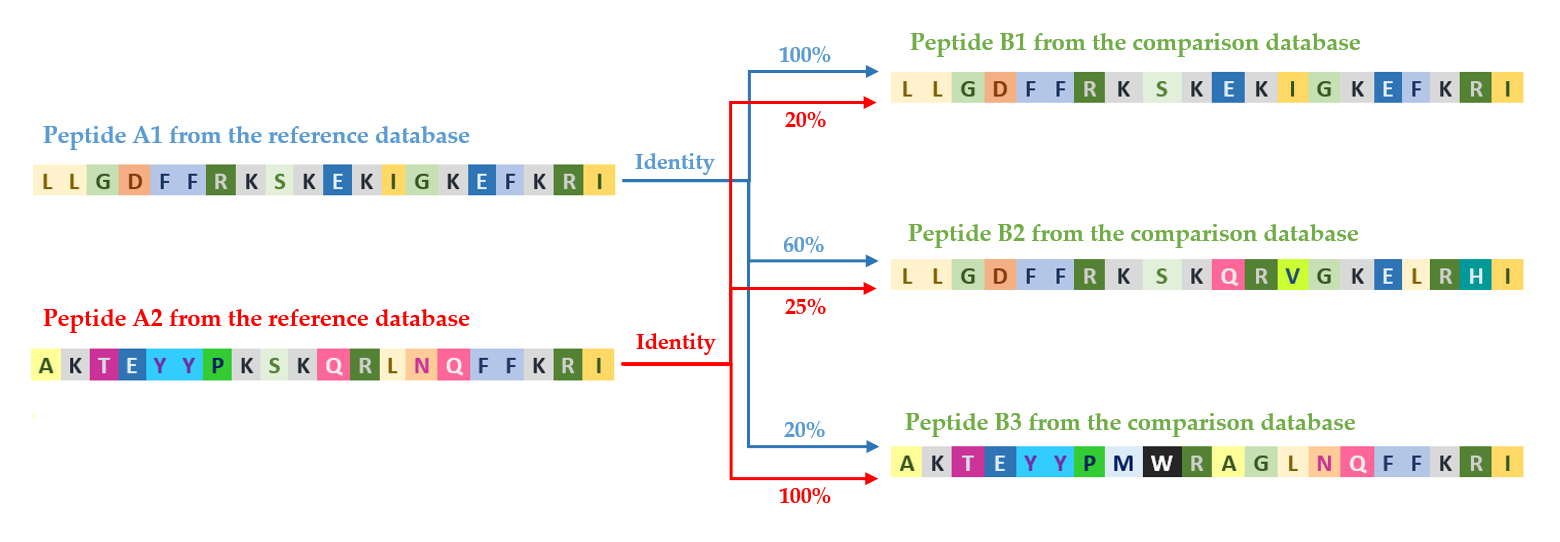
The figure shows how sequences A1 and A2 from the reference database are compared with sequences B1, B2, and B3 from the query database, resulting in the determination of a percentage identity for each pair.
Key Features
What makes diamond a useful tool in bioinformatics?
Exceptional Speed
Up to 20,000 times faster than BLASTX, allowing for the analysis of huge datasets on standard servers.
High Sensitivity
The use of a reduced amino acid alphabet and "spaced seed" technology ensures high sensitivity with minimal loss of precision.
Wide Application
Ideal for proteome analysis, metagenomic projects, and identifying candidates for antimicrobial peptides.
Bibliography
Buchfink, B., Xie, C., & Huson, D.H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12, 59–60. https://doi.org/10.1038/nmeth.3176
Buchfink, B., Reuter, K., & Drost, H.G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods, 18, 366–368. https://doi.org/10.1038/s41592-021-01101-x
Marczak, B., Bocian, A., & Łyskowski, A. (2025). Antimicrobial Peptide Databases as the Guiding Resource in New Antimicrobial Agent Identification via Computational Methods. Molecules, 30, 1318. https://doi.org/10.3390/molecules30061318